捉妖记 - 一个奇怪的插入片段长度分布图
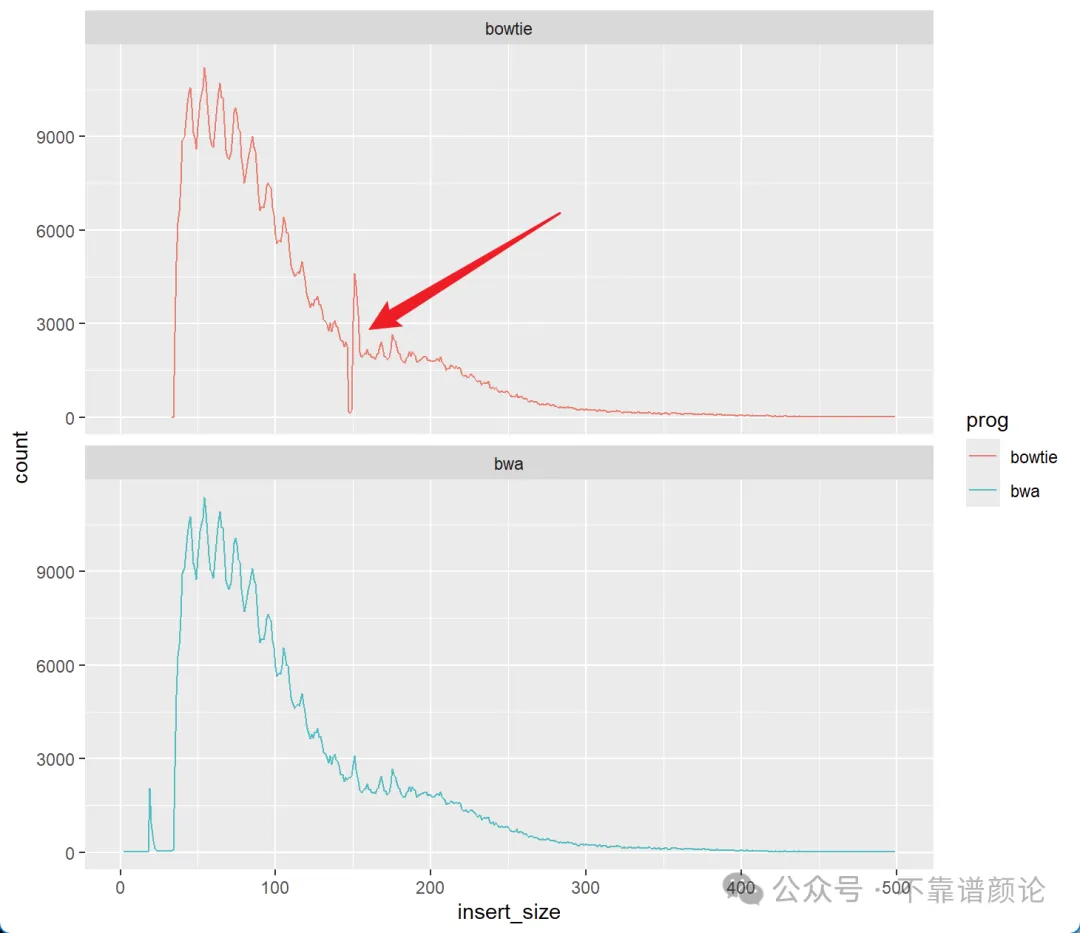
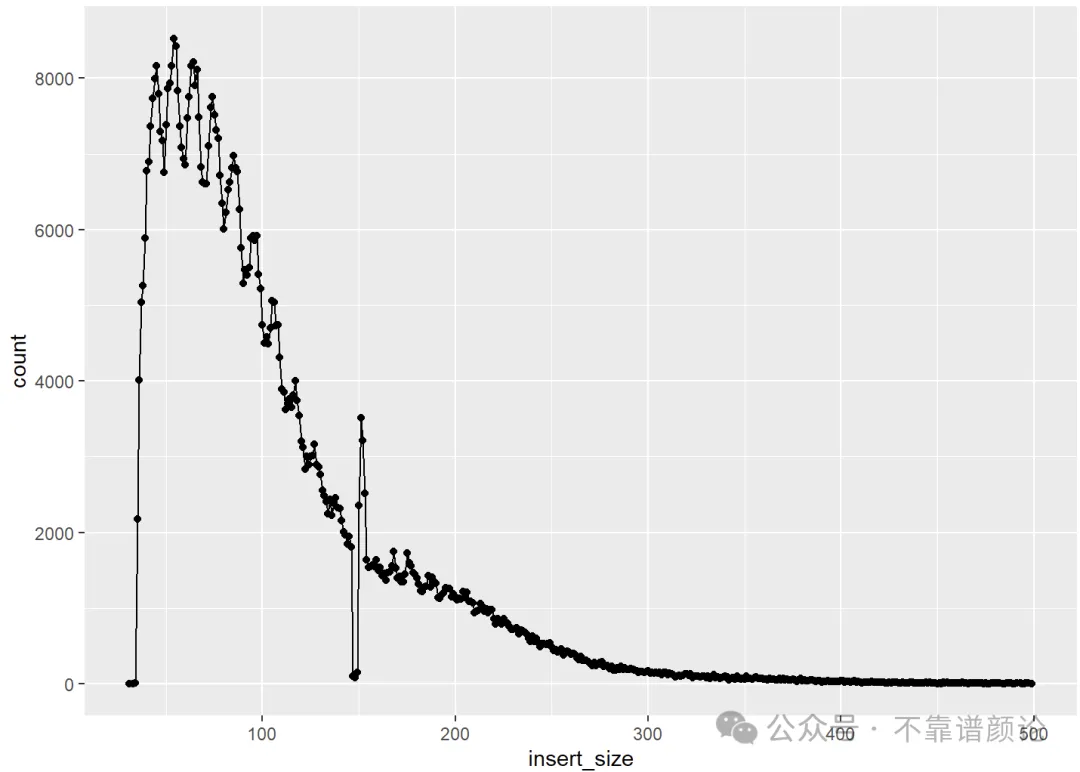
这是某个NGS数据的插入片段长度分布图(上图用的是 bowtie2 做的序列比对,下图是用的 bwa),在 bowtie2 的结果中,约150bp处有一个明显异常的突刺,看得人如鲠在喉。虽然它也许并不会严重影响后续分析,也经常就被大家睁一只眼闭一只眼地忽略了,但所谓“事出反常必有妖”,咱做生信分析的,偶尔遇到这样的“无伤大雅”的小妖,不妨瞪大眼睛仔细看看,或许会有意想不到的收获呢。
首先,我们来尝试从公共数据中重现该问题。
直接上代码(需要用到的软件、参考基因组及其索引等,可自行上网搜索解决,这里不花篇幅赘述):
|
|
然后,到 R 语言环境中绘图:
|
|
至此,我们就得到了题图中的那个奇怪曲线。
接下来,开始捉妖:
-
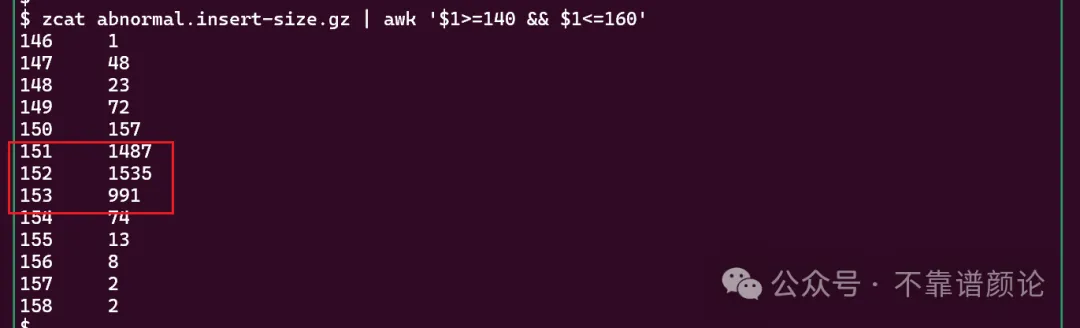
先从用来画图的 insert-size 数据开始查看:
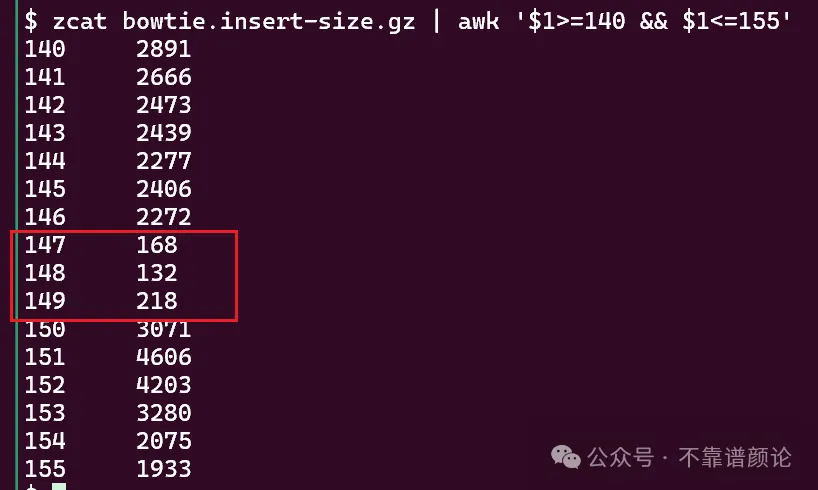
原来出问题的是 147 ~ 149 这连续的三个数据点,相比周围明显偏低(其实还包括后续 150 ~ 153 这四个数据点,它们是相比周围明显偏高)。
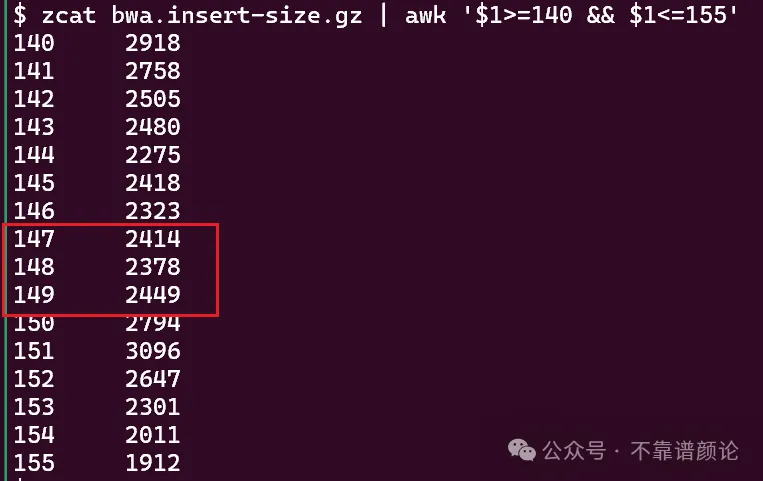
然而,在 bwa 的结果中,并没有类似情况发生。这意味着,bwa 的结果中,片段长度为 147 ~ 149 的这些 read pairs,有相当一部分,在 bowtie2 的结果中,片段长度变成了别的数值(变大或变小)。
-
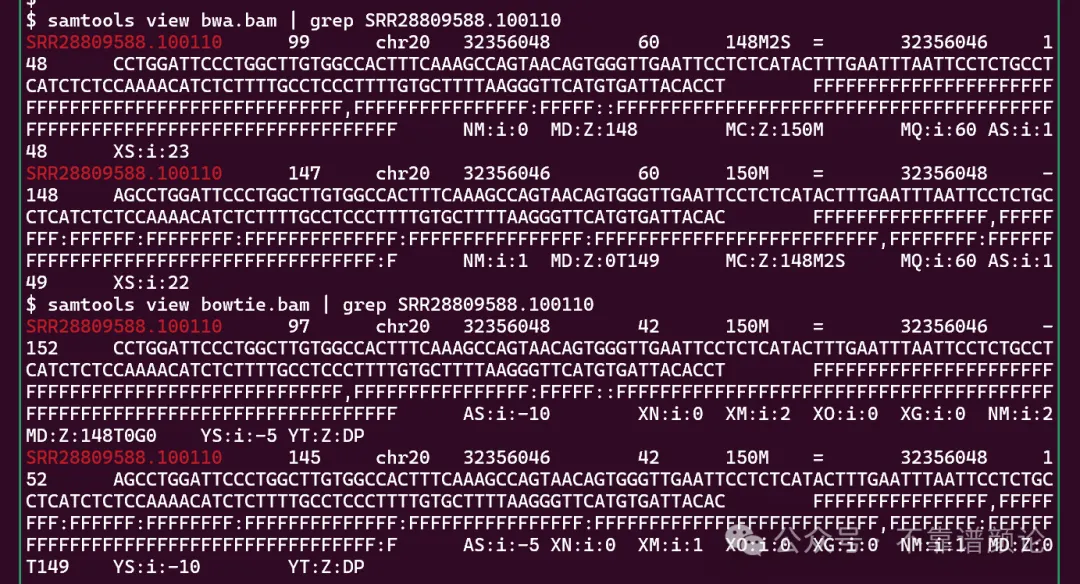
接下来,我们就从 bwa 结果中,提取这些 read 名称,到 bowtie 结果中把它们的长度提取出来:
1 2 3 4 5 6 7 8 9 10samtools view bwa.bam \ | awk -F'\t' '$9>=147 && $9<=149{print$1}' \ > read-names.txt samtools view bowtie.bam \ | grep -F -w -f read-names.txt \ | awk '$9>0{s[$9]++}END{for(i in s){print i"\t"s[i]}}' \ | sort -k1,1n \ | gzip -9 \ > abnormal.insert-size.gz继续用 R 画出来看看:
1 2 3 4 5 6 7 8a3 = read_tsv("abnormal.insert-size.gz", col_names = c("insert_size", "count"), col_types = "ii") a3 %>% ggplot(aes(insert_size, count)) + geom_line() + geom_point()结果呢:
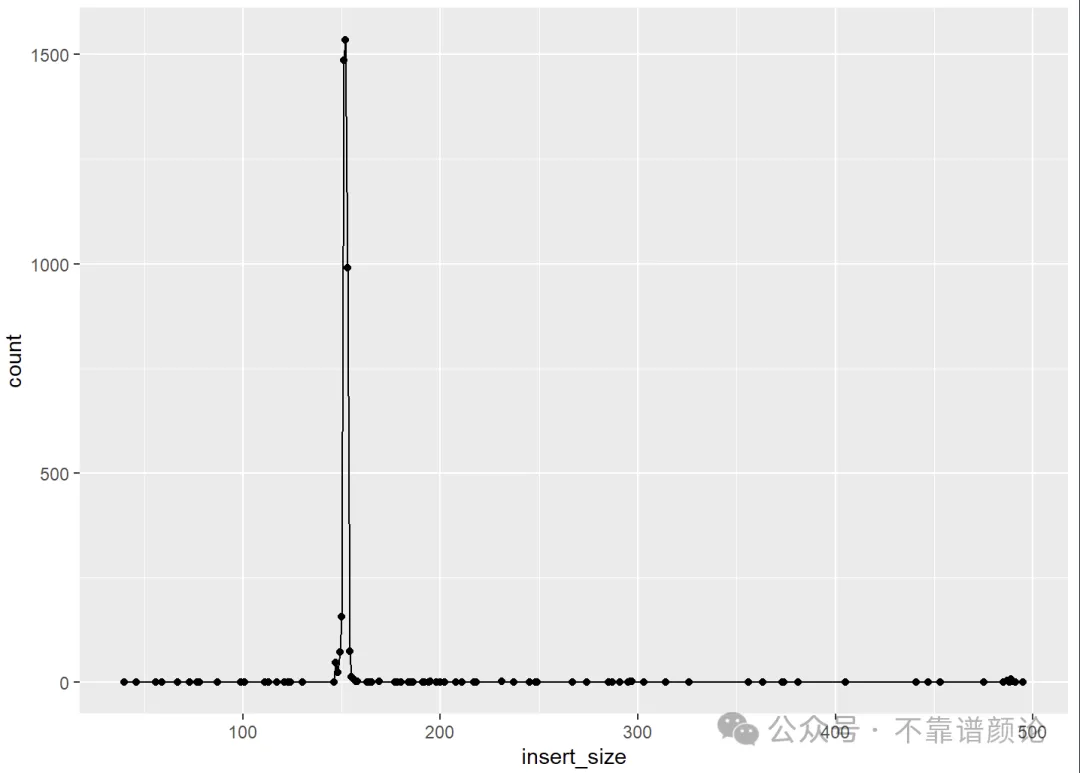
各种长度都有,这还真是意想不到呢。
-
既然这样,我们就先选择相对最极端的情况来看看。
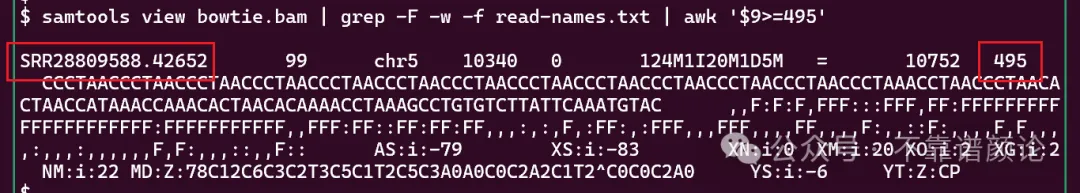
这对 read pair,名为 SRR28809588.42652,在 bowtie2 的结果中,片段长度为495。我们可以分别在 bowtie2 和 bwa 的结果中,搜索该 read name。
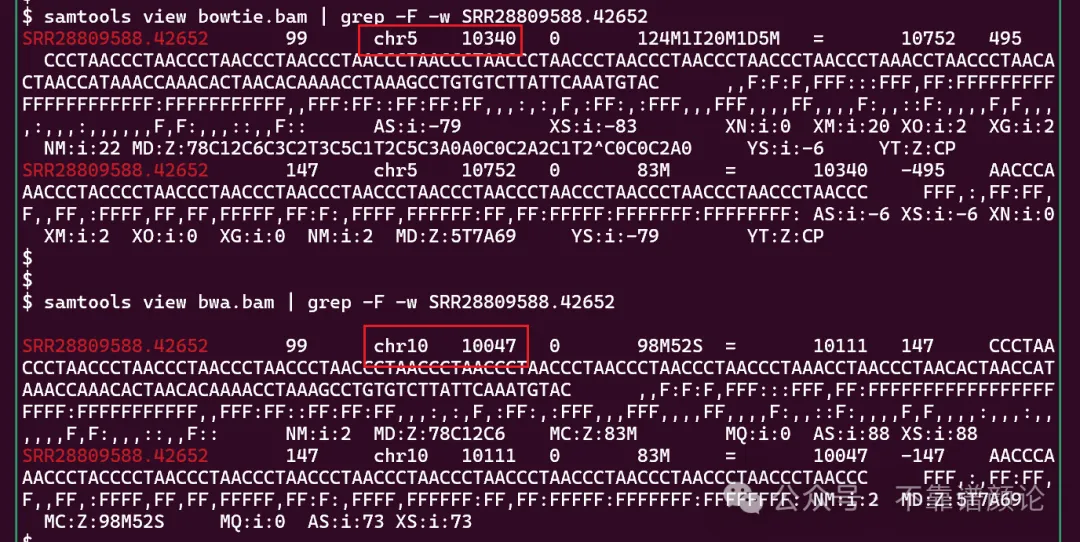
它们竟然分别比对到了 chr5 和 chr10,比对的 position 数值很小(10340 和 10047),这明显在染色体的端部。而序列呈现,CCCTAA 的串联重复,这是端粒重复序列,也就难怪它会被“随机”地比对到不同染色体去。
-
继续往下追查,可以发现它们都跟短串联重复序列相关,导致同一 read pair 被不同工具比对到了不同基因组位置,自然呈现出了差别较大的插入片段长度。
于是,很自然地,可以考虑,通过使用将这些串联重复序列 mask 掉的 fasta 序列作为参考基因组,是否能规避这个问题呢?
从 UCSC 的 goldenPath 上下载该文件试试:
http://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/
(此处省略若干步骤,过程基本同前)
然而,事实上,问题并没有得到解决。
-
这个 150bp 在数值上与测序的读长(PE150)很像,不妨做个小实验,把读长一开始截短至 100bp,重新跑一遍此前的分析试试。
1 2 3 4 5 6 7 8 9 10 11zcat SRR28809588_1.fastq.gz \ | head -n 4000000 \ | cut -c1-100 \ | gzip -9 \ > test2.1.fq.gz zcat SRR28809588_2.fastq.gz \ | head -n 4000000 \ | cut -c1-100 \ | gzip -9 \ > test2.2.fq.gz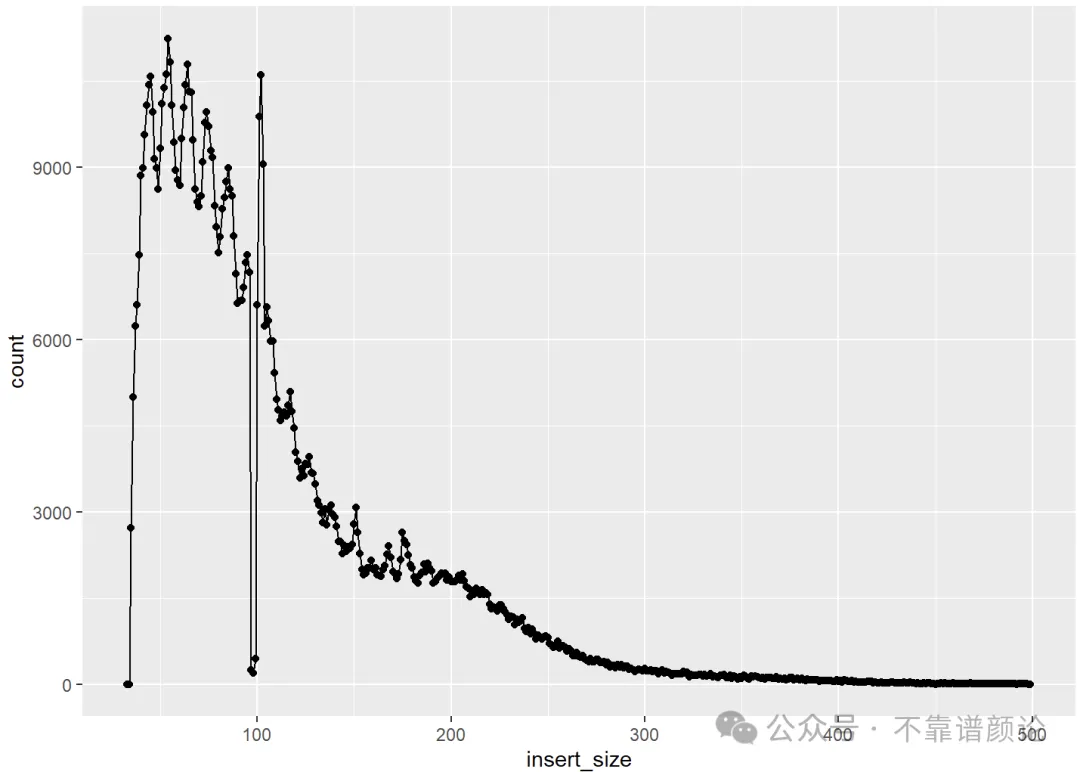
曲线上异常的突刺果然乖乖左移到了 100bp 处,看来这个问题的确是跟原始测序reads的读长有关。
-
再仔细观察异常数值附近:
主要的片段长度变化,确实只是从原来的 147 ~ 149 变成了 151 ~ 153,增加大约 4bp。那么,接下去,我们就针对这种 “片段长度增加了 4 bp” 的特征,去找出典型的片段,观察其是否存在某些规律,以便揭示其发生的原因。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17# 从 bwa 结果中提取 reads 的名字和片段长度 samtools view bwa.bam -F 0xF04 \ | grep -F -w -f read-names.txt \ | awk '$9>0{print$1"\t"$9}' \ | sort \ > bwa-align.txt # 从 bowtie2 结果中提取 reads 的名字和片段长度 samtools view bowtie.bam -F 0xF04 \ | grep -F -w -f read-names.txt \ | awk '$9>0{print$1"\t"$9}' \ | sort \ > bowtie-align.txt # 关联两个结果文件,并筛选出差值刚好为 4 的记录 join -j 1 bwa-align.txt bowtie-align.txt \ | awk '$2+4==$3' | less -S至此,我们就拿到了这些片段的名称,可以逐个进行检查。
此时,就需要把 bam 文件排序并建立索引,以便加载到 IGV 中进行查看了(可交互的可视化工具相当重要呢)。
1 2 3 4samtools sort bowtie.bam > bowtie.sort.bam samtools index bowtie.sort.bam samtools sort bwa.bam > bwa.sort.bam samtools index bwa.sort.bam下面展示其中一个典型结果在 IGV 中的表现:
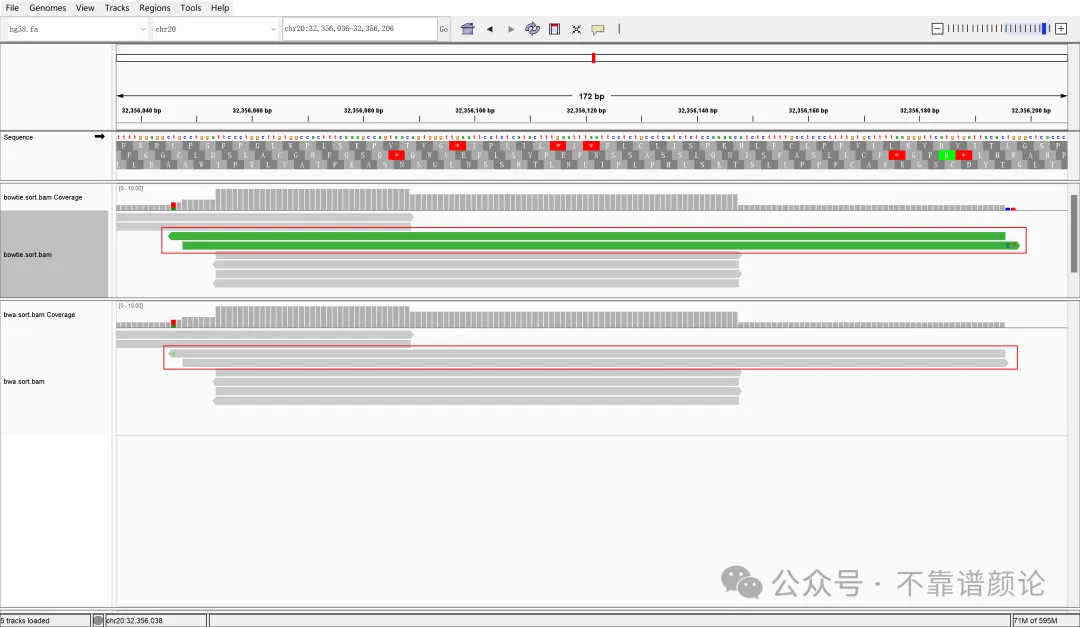
将两端放大来看:
左侧端:

右侧端:
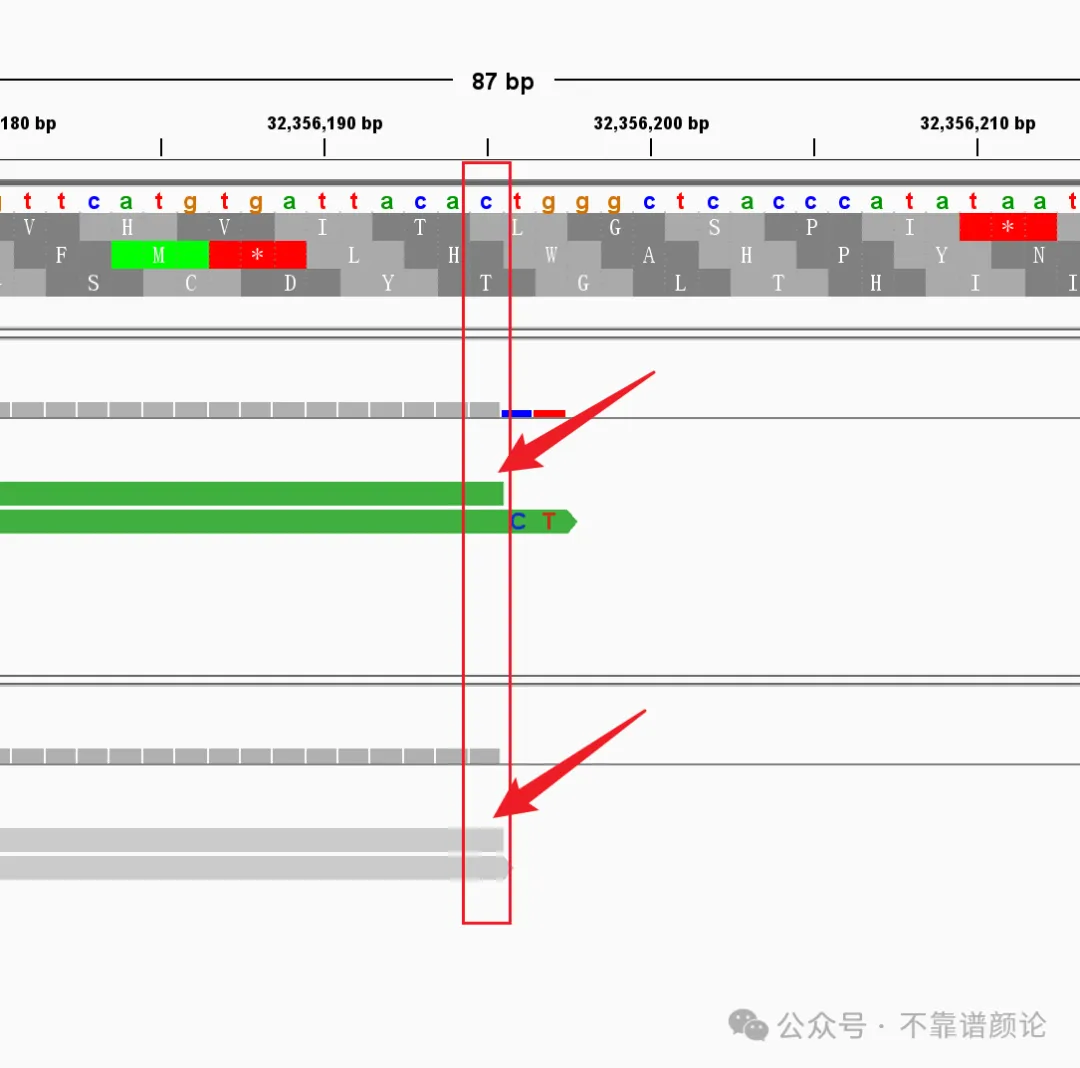
可见,这一对 read pair(SRR28809588.100110),其片段左端位于 chr20 的 32,356,048 位置,右端位于 32,356,195 位置,片段长度应为 (32,356,195 - 32,356,048 + 1) = 148bp。
然而,因为双端测序的每一端都测了 150bp,于是两端都会有 2bp 测穿,形成了比对结果中的 soft clip。在 bwa 的结果中,这个 soft clip 被正确计算,得到了 148 的片段长度,写入了 BAM 文件的第九列,而 bowtie 的结果,明显是没有正确处理该信息,仍然按照比对 CIGAR 的 150M,给出了错误的信息。
至此,真相大白,即在刚好在 read 末端存在少数碱基的 soft clip 时,bowtie2 的程序对片段长度的计算存在 bug。所幸这个问题并不影响其比对结果,所以下游分析并不会因此受到影响(严重依赖该 BAM 文件中的第九列的情况除外)。
捉妖结束,收工。关于是否可能通过流程中命令参数的调整,来解决这个问题,欢迎大家留言补充。
注:本文首发表于“不靠谱颜论”公众号,后同步至本站。