ARTS第一周(2019年12月16日~22日)
Algorithm
全局比对(Global Alignment)
算法简介
准确地说,这叫双序列全局比对算法(Pair-wise Global Alignment Algorithm),也叫做Needleman-Wunsch算法,于1970年由Saul B. Needleman和Christian D. Wunsch提出。这是生物信息学中最经典的算法,也几乎是所有介绍生物信息学算法的起点,因为所有后续的各类序列比对,基本上都是由这个算法衍生出来的。也因此,我在反复挑选之后,终于还是用它来作为第一次打卡的主题。
这个算法,包括后来衍生出的各种序列比对算法,其核心都是动态规划(Dynamic Programming),后者的基本思想是,一个问题可以被拆分成一系列子问题,从这些子问题的解决方案中,就可以选出解决原问题的最优化解决方案来。
详细介绍本算法的文章很多,我这里就不做进一步展开,只引用来自wikipedia的一张图示如下:
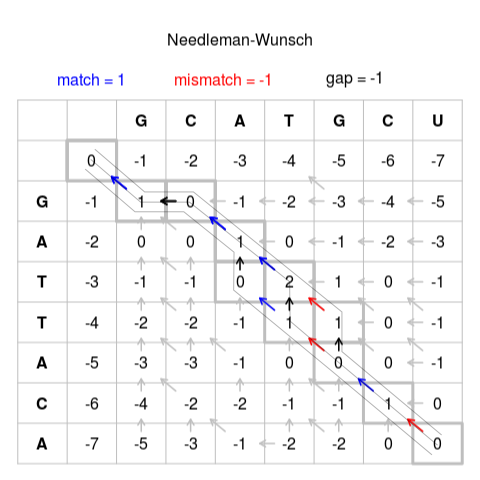
这个图,把两条序列之间的比对关系,从一个复杂抽象的组合概念,映射到了一个二维的矩阵上,成了一个容易图示的光标在矩阵上的游走,且整个矩阵,正好枚举了所有可能的比对关系组合。此后,每个方格都来自左上、左、上三个方格,分别经过match/mismatch、insertion、deletion而来,只要给定相应的打分规则,就可以从中选出最优的方案记下来,成为当前方格(子问题)的最优方案,进而一步步得出最终整个问题的最优化解决方案,即两条序列完整的比对结果。整个算法的时间复杂度和空间复杂度都是O(nm)。
我的实践
我选择这个算法开篇的另一个原因是,刚好前几天leetcode刷题上也用到类似思路。这是leetcode上的i第44题《通配符匹配》,给定一个字符串(s)和一个字符模式(p),字符模式中允许使用?表示任何单个字符,*表示任意字符串(包括空串),要求判断是否匹配(比如:“aa"与"a"不能匹配,而"aa"与”*“能匹配)。
对这道题,我首先想到的做法是回溯法(详见我的解法-1),根据规则挨个字符进行评判,如果遇到不匹配的,就往前回溯,让“*”在回溯过程中分别匹配不同长度的字符。这个算法逻辑上没问题,但最终提交时报出了“运行时间超出限制”。
考虑到这是一个典型的全局比对过程,于是我将其改为动态规划实现(详见我的解法-2),于是程序运行效率得到大幅度优化,提交通过。由于该题只需要判断是否匹配,因此我甚至不需要在内存中保存整个nxm大小的矩阵,而是按行扫描,只需要两个n长度(字符串s的长度)的数组即可。
算法的意义
序列比对是生物信息学中最重要的算法,几乎无所不在。序列比对的结果,一方面能给出两条序列的相似程度的评价,从而可以给出不同序列之间的“亲疏”关系,另一方面能给出比对细节,从而能够理解序列之间通过突变发生的可能的“演化”关系。有了这样的评价方法,后续的各种研究,才有了量化的依据和相应的实现基础。
参考
Resource
ClinVar
简介
ClinVar发布在NCBI上,其地址是:https://www.ncbi.nlm.nih.gov/clinvar/
这是一个收录了人类突变及对应表型信息的公开数据库。其信息,为各类基因检测的结果解读,提供了非常重要的支持。
ClinVar接收来自各类临床研究的数据提交,这些数据包括病例的临床表型信息、基因信息以及其他相关信息。
当前发展
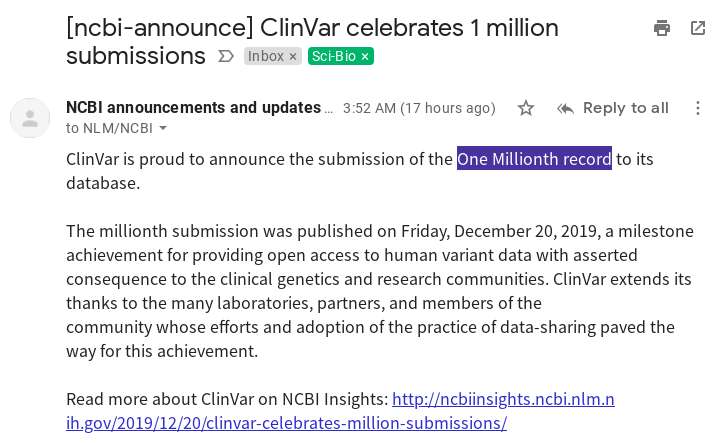
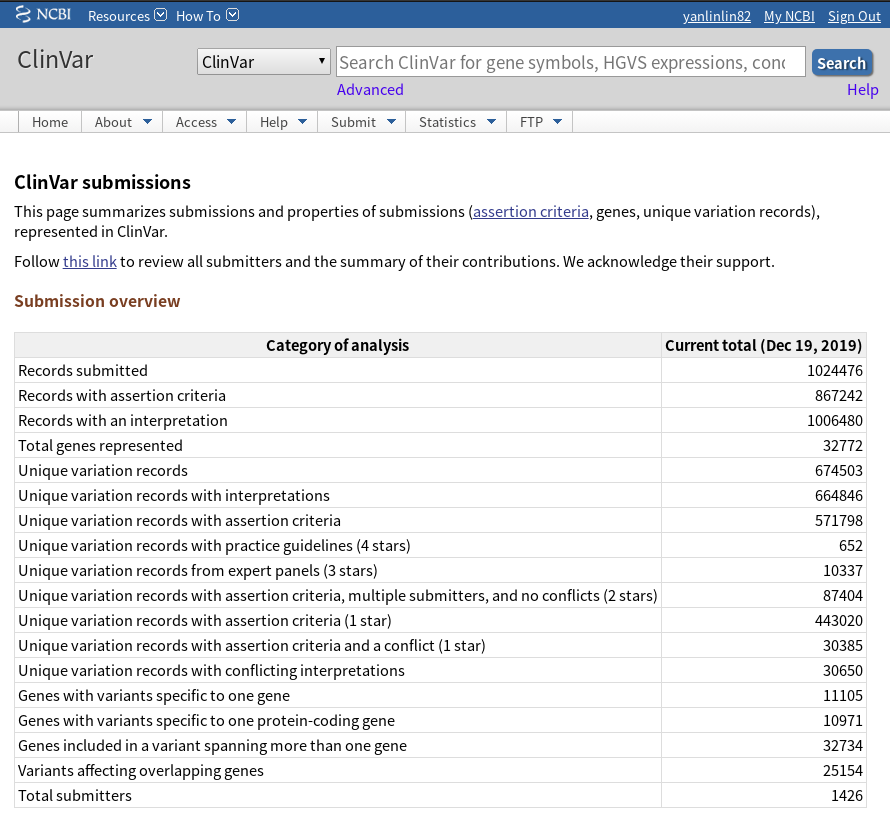
最近两天,该数据库的记录数也突破了百万,我截了两个图,展示如下:
参考
Technology
器官芯片化(OoCs,Organs-on-Chips)
这项技术,将组织器官放到芯片上,在体外模拟体内环境,从而用于相关的科学研究。目前将多种组织器官整合到一个体系中,以期用于疾病建模和药物发现等领域。《Trends in Biotechnology》上最新的这篇综述,对这个方向的技术进展做了总结。